MCCE Mechnism
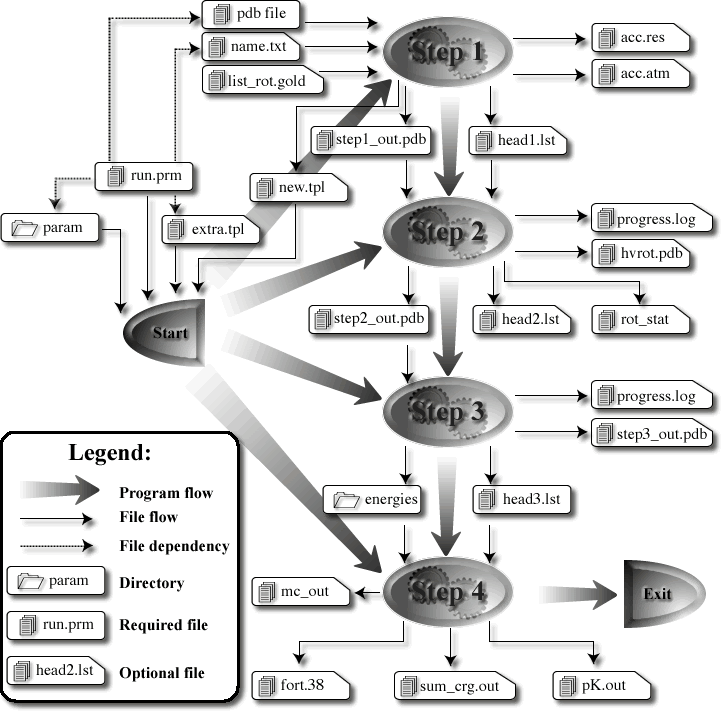
A MCCE run is a 4-step procedure
- Step 1 is file formatting,
- step 2 is rotamer making,
- step 3 calculates energy look-up table,
- step 4 is Monte Carlo sampling of conformers at each pH or Eh.
MCCE program can run any steps providing the prerequisite files exist in the working directory. Files required and written out by the program are illustrated in this chart. This file flow chart shows the summary of file dependencies.
Step 1, Formatting pdb file
Input files:
-
-
pdb file: input structure file in pdb format
-
name.txt (optional) : residue and atom renaming rule
-
list_rot.gold (optional): hot residue spot definition
-
Output files:
-
-
acc.res: solvent accessibility of residues
-
acc.atm: solvent accessibility of atoms
-
new.tpl (not always created): parameter file template of unrecognized cofactors
-
head1.lst (optionally used by step 2): summary of rotamer making policy of residues
-
step1_out.pdb (used by step 2): step 1 output file is in mcce extended pdb format
-
Step 1 prepares an extended pdb file suitable to be read into step 2. The input pdb file is in standard pdb format. It can have alternative side chain positions, but mcce can not process alternative backbone positions. Alternative side chains are treated as side chain conformers. When side chain atoms are missing, mcce will complete the side chain atoms at the torsion minimum. In this step several things will happen:
-
-
With the instructions in the renaming rule file "name.txt", residues and atoms will be renamed so that a cofactor can be split into several independent ionizable groups (for example, heme can be divided into heme and two propionates) and several groups can be combined as one (for example, heme can be grouped with the axial ligands).
-
Step 1 will identify unrecognized cofactors and interpret them as non-charged atom assemblies.
-
This step completes the missing atoms in each known residue (note: If your file has individual residues with missing atoms the tpl file for that residue will be used to add missing atoms).
-
This step calculates the solvent accessible surface (SAS) area and strips off exposed water and salt (HOH, NO3 and SO4). The SAS threshold of stripping off water and salt is defined by $(H2O_SASCUTOFF in run.prm or as user defined parameter in step1.py command).
- This step gives warnings on geometry clashes between atoms which are not supposed to be bonded.
- This step identifies heme ligands and cys-cys disulfide bridge.
-
This step extracts N terminus and C terminus of a chain.
-
The renaming rule file "name.txt" instructs the mcce program to rename atom name, residue name, sequce number, and chain ID. Here is several sample lines in this file:
# Symbol "*" in the first string is a wildcard that matchs any character.
# It means "do not replace" in the second string.
#
# The replace is accumulative in the order of appearing in this file.#
*****HEA****** *****HEM******
*****HEC****** *****HEM******
*CAA*HEM****** *****PAA****** extract PAA from heme
*CBA*HEM****** *****PAA******
*CGA*HEM****** *****PAA******The line started with "#" and the line shorter than 30 characters are comment lines. For other lines, the first 30 characters should be two 14-character strings separated by exactly two spaces, and the rest of the line is comment field. A valid line instructs mcce program to replace string 1 by string 2. The mcce program will match this string with position 13 to 26 of a coordinate line in the input pdb file. The symbol "*" is the wild card that matches any character in strings. The replace action is accumulative and order sensitive. For example, The line
HETATM 1683 CAA HEC 1 1.317 -3.987 -1.685 1.00 0.00 C
will be renamed to
HETATM 1683 CAA HEM 1 1.317 -3.987 -1.685 1.00 0.00 C
then
HETATM 1683 CAA PAA 1 1.317 -3.987 -1.685 1.00 0.00 C
Another file step 1 may use is "list_rot.gold". This file defines "hot spots" in a protein where rotamers will be made with a step of 30 degrees in step 2. If a coordinate line of any atom of a residue is present in this file, this residue and residues within 4 angstroms will be flagged to be "hot spots" in file "head1.lst", which will be used by step 2.
The output files of step 1 "acc.res" and "acc.atm" contain the solvent accessible surfaces of residues and atoms. In "acc.res", both absolute value and percentage of the solvent accessibility are listed.
When unrecognized cofactor is encountered in step 1, a parameter file will be created with name as "new.tpl" and a warning message will be issued. The atom connectivity is guessed and all atoms are assumed to have charge 0. This file can be the starting point of making a parameter for a new cofactor. If the program is resumed from step 2 or 3, this file will be read in byto stepdefine 0the asmolecule a parameter file so step 2 and 3 can proceed without step 1.parameter.
The file "head1.lst" lists the rotamer making policy of the residues. When $(ROT_SPECIF) in "run.prm" is set to "t", this file will be used as instruction of rotamer making of step 2, otherwise, file "head1.lst" will be ignored. This file can be modified and will be effective if the program resumes from step 2. The rotamer making is also dependent on step 2 rotamer making level. If step 2 is running at level 1 (quick run), the rotation rotamers will not be made even though head1.lst rules say say. The behavior will change in the planned new version of mcce.
The file "step1_out.pdb" is a formatted pdb file, which will be read in by step 2. The mcce extended pdb format contains 3 more fields than standard pdb file. Right after atom coordinates, these three fields are charge, size and rotamer making history.
Step 2, Making rotamers
Input files:
-
-
step1_out.pdb: input structure file of step 2 in mcce extended pdb format
-
head1.lst (optional) : rotamer making policy of residues
-
Output files:
-
-
progress.log: progress report file, dynamically updated
-
rot_stat: rotamer making statistics, dynamically updated
-
hvrot.pdb: heavy atom (without hydrogen atoms) rotamer pdb file
-
head2.lst (optionally used by step 3): summary of rotamers made in step 2
-
step2_out.pdb (used by step 3): step 2 output file with multiple rotamers in extended pdb format
-
Step 2 makes and optimizes rotamers from the structure in "step1_out.pdb". In this step, the rotatable bonds (defined in parameter files) of each residue is rotated by the steps defined in "run.prm". Then the self van der Waals (VDW) potential (interaction among atoms of the same side chain excluding 1-2 and 1-3 interactions, and interaction between the side chain and backbone atoms) is calculated. Side chain rotamers with high self VDW potentials are deleted. Then the side chains are optimized with possible hydrogen bond partners. A number of repackings starting from randomized initial structures (one conformer from one residue) are performed to reduce side chain rotamers to those with low energy local packings. Ionization states are then created and protons are placed on side chains. At the end of side chain rotamer optimization, MD simulations are carried out locally to relax the structure.
The input file step1_out.pdb is the only essential file step 2 will use. You can modify this file to add, delete or edit residues without causing problems as long as the file observes mcce extended pdb format. Sometimes a little editing of this file is necessary, for example, the terminal residues are not always correctly identified by the mcce program due to "broken chains" caused by the missing residues in the pdb file.
The file "head1.lst" provides residue specific rotamer making rule. It will be used only when $(ROT_SPECIF) is set to be "t". The line of this file such as:
NTR A0003_ R t 06 S f 0.0 H t 06 M 000
is interpreted as "Rotate is true and 6 steps per bond, then swing is false (angle is 0 if any), then Hydroxyl relaxation is true and the maximum number of starting conformers per hydroxyl is 6, and the maximum total number of the conformers is not limited". If you want to investigate a specific site in details, change the step 6 to 12. But making 12 steps for many sites (>30) are not recommended because it is expensive in terms of memory and CPU time.
The file "progress.log" is a dynamically updated progress report file. It lists mainly the repacking progress.
The file "rot_stat" is an important file to review the rotamer making history. This file lists the number of rotamers of each residue. It is easy to tell what residues get rotamers and if the total number of rotamers is manageable (a 2000 conformer structure may need one day to run the next two steps, step 3 and 4).
The file hvrot.pdb is a file at the same format as step1_out.pdb and step2_out.pdb but lacks hydrogen atoms. The main use of this file is to rename it to "step1_out.pdb" and run step 2 again with "swing" rotamers instead of "rotate" rotamers. This provides a way to relax the structure by "swinging" the rotatable bond a little and reevaluate the rotamers. This feature is most for advanced users to calibrate hydrogen bond directed rotamer making algorithm and MD relaxation subroutine.
The file "head2.lst" is a summary of the rotamers made in step 2. This file is not used by step 3.
The file "step2_out.pdb" is in the mcce extended pdb format. It is the single file connecting step 2 and step 3.
Step 3, Calculating the energy lookup table
Input files:
-
-
step2_out.pdb: input structure file of step 3 in mcce extended pdb format
-
Output files:
-
-
progress.log: progress report file, dynamically updated
-
energies (directory of energy lookup table used by step 4): opp files, each opp file is pairwise interaction to a conformer
-
head3.lst (used by step 4): list of conformers and self energy of conformers. It is used by step 4.
-
step3_out.pdb: step 3 output file with multiple rotamers in extended pdb format
-
Step 3 calls PBPoisson Boltzmann equation solver, DelPhi, to calculate reaction field energy and electrostatic pairwise interaction. The result is stored as together with van der Waals interactions as one file per conformer. These files have extension "opp" and are located under directory energies. The self-energy terms (not dependent on side chains of other residues) of conformers are listed in file "head3.lst" The progress is dynamically updated is file "progress.log".
The file "progress.log" reports the progress of DelPhi calculation, which can be used to estimate the total time of this step. There will be two parts of DelPhi calculations: the first is pairwise calculation and the second part is the reaction field energy calculation. It takes less time on reaction field energy calculation than on pairwise calculation.
The directory "energies" holds the pairwise conformer interaction lookup table as files with extension '.opp' and starting with the conformer_id, e.g. GLU02B0012_002.opp.
These files are header-less, but following is each column description:
column #: 1 2 3 4 5 6
description: conf# name corr_el vdw_pwise delphi_el post_bdry_corr_el (kcal/mol)
The file "head3.lst" contains self energy of each conformer and control flags of step 4. The flag is: "t" for fixed occupancy 0 or 1, or "f" for free to sample. The energy unit is Kcal/mol.
The file "step3_out.pdb" is an extended pdb file with multiple conformers. The conformer number is sorted to be continuous and consistent with the conformer numbers in file "head3.lst" and step 4 output file fort.38. This file is identical to "step2_out.pdb" if "step2_out.pdb" is an unmodified file created by step 2.
Step 4, Simulating pH or Eh titration with Monte Carlo sampling
Input files:
-
-
energies (directory): energy lookup table for pairwise interaction between conformers
-
head3.lst: self-energy of conformers and Monte Carlo sampling flags of conformers
-
Output files:
-
-
mc_out: progress of Monte Carlo sampling and energy tracing
-
fort.38: conformer occupancies
-
Step 4 is a titration simulation by Monte Carlo sampling. The Monte Carlo sampling is performed at specified set of pH/Eh. At each titration point, there will be several (predefined in "run.prm", the default is 6) independent samplings. Each sampling goes through annealing, reducing, and equilibration stages. Statistics of conformer occupancy is only done at equilibration statge. Yifan's Monte Carlo subroutine will check early convergence and quit sampling early to save time. The result is reported as conformer occupancy in file "fort.38".
The file "mc_out" is the progress report of Monte Carlo sampling. It contains running energy tracing which can be used to calculate the average E or enthopy of the system, or verify if the system is trapped at local energy minima. By "grep Sg mc_out", you can find the standard deviation of independent samplings.