How to add rotamer making rules to parameter file?
How to add rotamer making rules to parameter file?
If a residue or a cofactor has rotatable single bond, the user needs to define the rotation rule so that MCCE can make rotamers accordingly.
In MCCE, the rotation is for heavy (non H) atoms only, and H atoms have their own position optimization rules.
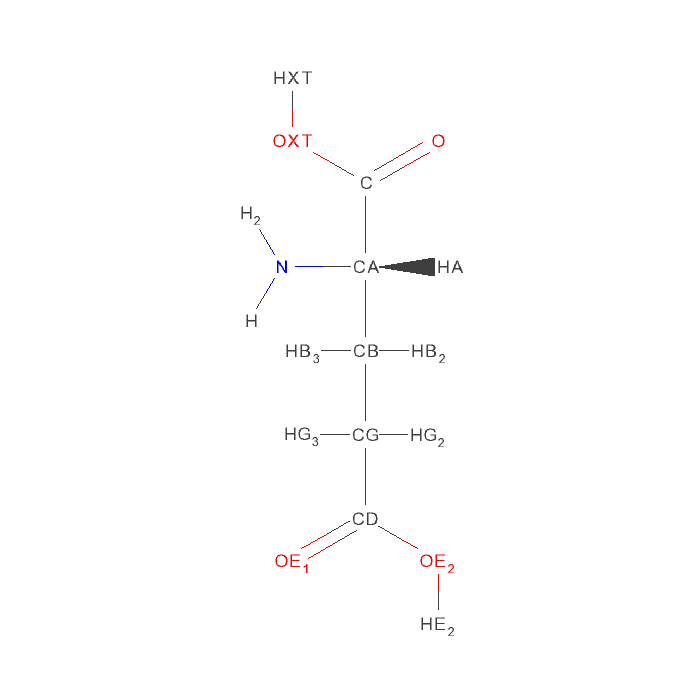
Let's look at glutamic acid (GLU). Its backbone atoms N, C, CA are considered "fixed". The side chain atoms CB, CG, CD, OE1 and OE2 are susceptible to rotationo. Here is how the rotation rules are defined in glu.ftpl file.
# Rotatable bonds. The atoms extended in the bond direction will all be rotated.
ROTATE, GLU: " CA " - " CB ", " CB " - " CG ", " CG " - " CD "The first part, which is before ":" sign, is the parameter key "ROTATE, GLU". It means this line is rotation rule for GLU residue.
The second part, which is after ":" sign, is the parameter value " CA " - " CB ", " CB " - " CG ", " CG " - " CD ". The atom names are in quotes because the atom names in PDB format are 4-char including spaces. The parameter value defines 3 rotatable bonds, and each rotatble bond is defined by two atoms.
- The order of two atoms that define a rotatble bond is important. Follow the direction of the first atom to the second atom, all atoms connected to the second atoms will rotate around this rotatable bond.
- The order of rotatable bonds does not matter in MCCE rotamer making.
- The rotations resulted from multiple rotatble bonds are stackable, which means, rotations around one bond will be build on the rotations around another bond.
- All rotation rules of the same residue must be defined in a single ROTATE parameter line.