Gromacs Quick Start
GROMACS Quick Start
Official Documentation:
https://manual.gromacs.org/current/user-guide/index.html
Gromacs Server and Environment:
Gromacs is pre-installed on a Levich server and user accounts are initilized to include Gromacs path.
In case you want to see how the Gromacs environment was set, Here it is:
In file ~/.profile, the last line points to gromacs environment initialization:
source /usr/local/gromacs/bin/GMXRCUsers are expected to install their own Python and Python modules.
Install Conda Python and Python modules:
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.shchmod +x Miniconda3-latest-Linux-x86_64.sh./Miniconda3-latest-Linux-x86_64.sh (answer yes to initialize Minicoda3)Logout and back in to have conda environment, then run:
conda install numpy scipy matplotlib pandasConda can be deactivated or reactivated by command:
conda deactivateconda activateGromacs Command
Main Command:
gmkExamples:
gmx -h (print help)gmk -v (show version)gmx help [module] (documentation of a module)Example 2: Building Biphasic Systems
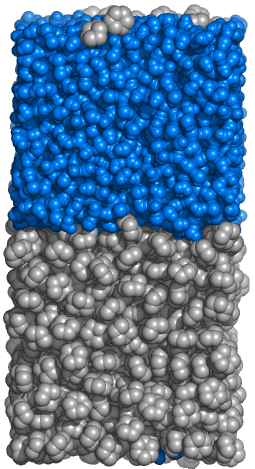
Make a heterogeneous biphasic system composed of hydrophobic (cyclohexane) and hydrophilic (water) layers.
This example illustrates the procedure of making a new molecule that the Gromacs doesn't know.