MCCE Commands
MCCE commands explanation and examples.
MCCE Program Run and Debug
Commands and Tools
getpdb
Command line tool to download pdb file from PDB
Syntax:
getpdb pdbID [file]
This program gets a pdb file from Protein Data Bank, and saves to a file with its PDB name or a user named file.
Example:
getpdb 1akk
Download with url https://files.rcsb.org/download/1akk.pdb.
Download completed.vdw_bk.py
Calculate conformer to backbone vdw (Lennard-Jones potential).
Syntax:
vdw_bk.py [-h] [-c cutoff] confID
Compute vdw1 breakdown at conformer level. The conformer ID can be found in head3.lst.
positional arguments:
confID Conformer ID as in head3.lst
optional arguments:
-h, --help show this help message and exit
-c cutoff Cutoff value of displaying conf vdw pairwise
Example:
(base) jmao@Jupiter:~/projects/1akk$ vdw_bk.py GLU-1A0090_005 -c 0.1
LEUBKA0064_000 -0.104
METBKA0065_000 -0.499
LEUBKA0068_000 -0.825
GLUBKA0069_000 -0.758
ILEBKA0085_000 -0.571
LYSBKA0086_000 -0.990
LYSBKA0087_000 -1.005
LYSBKA0088_000 -1.011
THRBKA0089_000 -0.876
GLUBKA0090_000 1.424
ARGBKA0091_000 2.383
GLUBKA0092_000 -0.204
ASPBKA0093_000 -0.113
LEUBKA0094_000 -0.120
Total -3.616vdw_conf2conf.py
Calculate conformer to conformer pairwise vdw (Lennard-Jones potential).
Syntax:
vdw_conf2conf.py [-h] [-c cutoff] [-v] confID confID
Compute detailed conformer to conformer vdw.
positional arguments:
confID Conformer ID as in head3.lst, two IDs required.
optional arguments:
-h, --help show this help message and exit
-c cutoff Cutoff value of displaying atom to atom vdw
-v Turn on verbose mode, displaying more details
This program calculates Lennard-Jones potential between a conformer pair, which should be consistent with the number in opp file under energies directory. The pair could be self to self, which is vdw0 term in head3.lst. The programs reports atom to atom interaction and atom connectivity as well.
The conformer ID can be found in head3.lst.
Example:
(base) jmao@Jupiter:~/projects/1akk$ vdw_conf2conf.py GLU-1A0090_005 THR01A0089_002 -c 0.01 -v
ATOM1 ATOM2 vdw dist cnct r1 e1 r2 e2 R_sum E_par
CB GLU0090A005 -> CB THR0089A002: -0.024 5.446 none 1.9080 0.1094 1.9080 0.1094 3.8160 0.1094
CB GLU0090A005 -> OG1THR0089A002: -0.010 6.361 none 1.9080 0.1094 1.7210 0.2104 3.6290 0.1517
CG GLU0090A005 -> CB THR0089A002: -0.016 5.883 none 1.9080 0.1094 1.9080 0.1094 3.8160 0.1094
GLU-1A0090_005 - THR01A0089_002: -0.150vdw_pw.py
Update Lennard-Jones potential of step 3 in files head3.lst and energies/*.opp
Syntax:
vdw_pw.py
This program updates Lennard-Jones potential of step 3 in
- file head3.lst (vdw0 and vdw 1) and
- files energies/*.opp (vdw column).
A copy of head3.lst will be made as head3.lst_bak and the copy of energies directory will be made as energies_bak. It corrects the some parameter issues in mcce step 3 and offers a chance to rerun vdw calculation without running PB solver again. Also it checks the possible inconsistancy in parameter files and comes with two other tools: vdw_conf2conf.py and vdw_bk.py to inspect the vdw interaction clashes.
Example:
vdw_pw.py
MCCE Data Analysis
MCCE data analysis tools
fitpka.py
Fit the titration curve of an ionizable residue.
Syntax:
fitpka.py [-h] RES [RES ...]
Fit a titration of charged residues
positional arguments:
RES Charged residue names to plot, as in sum_crg.out or pK.out
optional arguments:
-h, --help show this help message and exit
Required input file
- sum_crg.out
Example:
Find the residue IDs:
$ cat sum_crg.out
pH 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14
NTR+A0001_ 1.00 1.00 1.00 1.00 0.99 0.96 0.70 0.20 0.03 0.00 0.00 0.00 0.00 0.00 0.00
LYS+A0001_ 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 0.97 0.78 0.27 0.04 0.00 0.00 0.00
ARG+A0005_ 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 0.97 0.80 0.38 0.08
GLU-A0007_ -0.00 -0.01 -0.07 -0.38 -0.84 -0.98 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00
LYS+A0013_ 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 0.99 0.95 0.67 0.19 0.03 0.00
ARG+A0014_ 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 0.99 0.94 0.66 0.19
HIS+A0015_ 1.00 1.00 1.00 1.00 1.00 0.98 0.83 0.36 0.06 0.01 0.00 0.00 0.00 0.00 0.00
ASP-A0018_ -0.01 -0.10 -0.49 -0.89 -0.99 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00
TYR-A0020_ -0.00 -0.00 -0.00 -0.00 -0.00 -0.00 -0.00 -0.00 -0.00 -0.00 -0.01 -0.03 -0.09 -0.32 -0.73
ARG+A0021_ 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 0.99 0.91 0.59 0.22
TYR-A0023_ -0.00 -0.00 -0.00 -0.00 -0.00 -0.00 -0.00 -0.00 -0.01 -0.06 -0.32 -0.76 -0.96 -0.99 -1.00
LYS+A0033_ 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 0.97 0.78 0.27 0.05 0.01 0.00
GLU-A0035_ -0.00 -0.00 -0.01 -0.03 -0.12 -0.46 -0.88 -0.98 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00
ARG+A0045_ 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 0.99 0.91 0.56 0.21
ASP-A0048_ -0.03 -0.21 -0.69 -0.95 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00
ASP-A0052_ -0.00 -0.01 -0.07 -0.44 -0.84 -0.95 -0.99 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00
TYR-A0053_ -0.00 -0.00 -0.00 -0.00 -0.00 -0.00 -0.00 -0.00 -0.00 -0.00 -0.00 -0.00 -0.00 -0.00 -0.00
ARG+A0061_ 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 0.98 0.90 0.57
ASP-A0066_ -0.03 -0.20 -0.68 -0.93 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00
ARG+A0068_ 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 0.99 0.95 0.76 0.34
ARG+A0073_ 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 0.98 0.85 0.39 0.09
ASP-A0087_ -0.01 -0.08 -0.44 -0.88 -0.98 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00
LYS+A0096_ 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 0.96 0.76 0.33 0.08 0.02 0.01
LYS+A0097_ 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 0.98 0.85 0.40 0.08 0.01 0.00
ASP-A0101_ -0.00 -0.00 -0.01 -0.09 -0.51 -0.91 -0.99 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00
ARG+A0112_ 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 0.98 0.87 0.43 0.09
ARG+A0114_ 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 0.99 0.94 0.63 0.17
LYS+A0116_ 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 0.97 0.75 0.27 0.05 0.01 0.00 0.00
ASP-A0119_ -0.00 -0.00 -0.03 -0.19 -0.71 -0.96 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00
ARG+A0125_ 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 0.99 0.90 0.55 0.13
ARG+A0128_ 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 0.99 0.97 0.79 0.31
CTR-A0129_ -0.00 -0.04 -0.29 -0.75 -0.96 -0.99 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00 -1.00
----------
Net_Charge 18.93 18.34 16.22 13.47 11.05 9.70 8.68 7.57 7.01 6.38 4.53 1.82 -0.64 -4.61 -9.32
Protons 18.93 18.34 16.22 13.47 11.05 9.70 8.68 7.57 7.01 6.38 4.53 1.82 -0.64 -4.61 -9.32
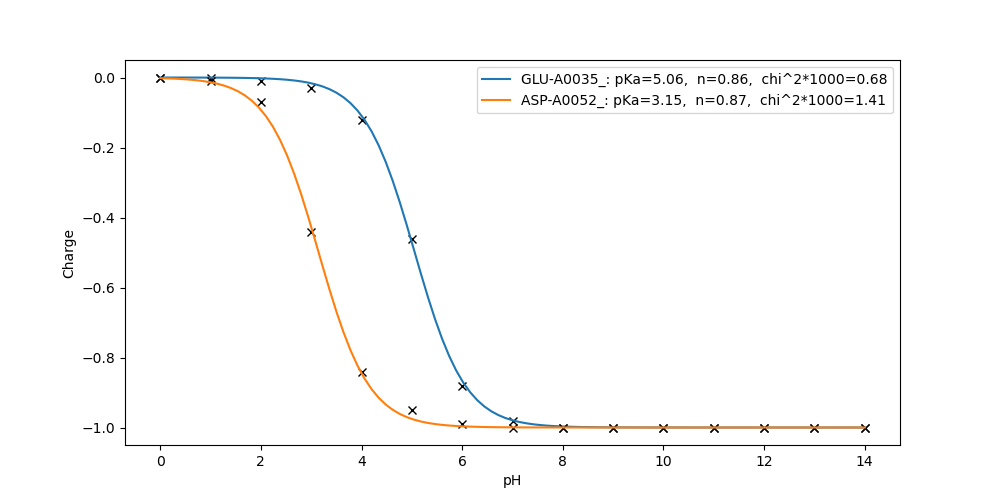
Electrons 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00Fit the titration curves of residue GLU-A0035_ and ASP-A0052_:
fitpka.py GLU-A0035_ ASP-A0052_
mfe.py
Analyze ionization free energy of a residue. It tells you why a residue has that pKa and what factors played a role.
Syntax
mfe.py [-h] [-p pH/Eh] [-x TS_correction] [-c cutoff] residue- residue: residue ID as in pK.out
- pH/Eh: pH value at which ionization energy is calculated. Default is the pKa (midpoint) where dG = 0.
- cut_off: Report only pairwise interaction bigger than this value.
- TS_correction: "t" to include entropy in G, "f" not to include, "r" (default) will look for run.prm. If entropy correction was turned on in step 4, mfe should not include this term as entropy has been "removed".
Required files:
- run.prm
- head3.lst
- extra.tpl for scaling factors that are not equal to 1.
- energies/*.opp for pairwise interactions
Example:
Check titration result in pKa.out:
cat pK.out
...
ASP-A0052_ 3.152
...
ASP-A0052_ is the residue_ID. Its calculated pKa is 3.152. At this point, the free energy of reaction from ASP neutral to ASP ionized should be close to 0.
Run
$ mfe.py ASP-A0052_ -c 0.1
Residue ASP-A0052_ pKa/Em=3.152
=================================
Terms pH meV Kcal
---------------------------------
vdw0 -0.01 -0.85 -0.02
vdw1 0.00 0.23 0.01
tors -0.10 -5.86 -0.14
ebkb -1.27 -73.44 -1.73
dsol 1.99 115.38 2.71
offset -0.62 -36.17 -0.85
pH&pK0 1.60 92.75 2.18
Eh&Em0 0.00 0.00 0.00
-TS 0.00 0.00 0.00
residues -1.36 -79.01 -1.86
*********************************
TOTAL 0.22 13.04 0.31 sum_crg
*********************************
ASNA0044_ -0.46 -26.92 -0.63 0.00
ARGA0045_ -0.11 -6.36 -0.15 1.00
ASNA0046_ -0.19 -11.09 -0.26 0.00
ASPA0048_ 0.50 28.96 0.68 -0.96
SERA0050_ 0.13 7.28 0.17 0.00
GLNA0057_ 0.22 12.78 0.30 0.00
ASNA0059_ -0.99 -57.18 -1.34 0.00
ARGA0061_ -0.26 -15.37 -0.36 1.00
ASPA0066_ 0.27 15.72 0.37 -0.94
ARGA0112_ -0.19 -11.11 -0.26 1.00
ARGA0114_ -0.10 -5.86 -0.14 1.00
=================================
You can do mfe calculation at pH other than mid-point.
MCCE Tools
List of miscellaneous tools MCCE offers for your research and convenience. Most of these tools support "-h" flags for additional information and use cases.
Some of these tools are intended for pre-run analysis, and some are intended for post-run analysis. Pre-run tools will be italicized, and are found in the MCCE_bin of a MCCE4-Alpha directory. Post-run tools will be bolded, and are found in MCCE4-Tools, a separate git directory.
cif2pdb (MCCE4-Tools)
usage: cif_to_pdb file.cif [file.pdb]
Converts a .cif file to .pdb format.
clear_mcce_folder (MCCE4-Tools)
Deletes all MCCE outputs from the present working directory, except: run.prm, the original PDB file, prot.pdb, and any non-MCCE files.
detect_hbonds.py
Detect H-bonds in a PDB file, with the option to include BK (backbone) atoms. hbonds_pdb_collection uses this function on a collection of PDB files.
usage: detect_hbonds.py [-h] [--include_bk] [--no_empty_files] [--out_dir OUT_DIR] [inpdb]
extract_md_frames
Extracts the trajectory's frames with the given indices into PDB files. Requires the MDAnalysis package.
filesdiff
Obtain the column difference between two MCCE files or the differences of all files in two MCCE output folders. Use the "-threshold" flag to output absolute differences beyond a given value (0 is default).
Applicable to the following MCCE files: 'all_pK.out', 'all_sum_crg.out', 'entropy.out', 'fort.38', 'head3.lst', 'pK.out', 'residues_stats.txt', 'sum_crg.out', 'vdw0.lst'.
fix_psf_mdanalysis
Provides a reformatted PSF file if "MDAnalysis" fails to parse the given PSF. Requires the "MDAnalysis" and "parmed" packages.
getpdb
Downloads one or more (bioassembly) PDB files from the RCSB Protein Databank. For example, to download triclinic hew lysozyme (4LZT), one could type in
usage: getpdb [RCSB protein code]
glossary
Gives detailed information regarding the various parameters of run.prm, where MCCE looks to handle more granular customization.
You can search for specific parameters by with a given (case-sensitive) prefix string. For example, "glossary T" will return all parameters starting with T, like "TITR_TYPE". The command "glossary --print" also prints the entire glossary.
hbonds_pdb_collection
Detects Hydrogen bonds, using detect_hbonds.py, over a collection of PDB files, in the step2_out.pdb format. ASK HOW TO USE THIS
usage: hbonds_pdb_collection [-h] [-input_dir INPUT_DIR] [-output_dir OUTPUT_DIR] [--include_bk] [--no_empty_files]
mcce_stat
Prints a table to keep track of progressing MCCE runs. Four "sentinel" files are looked for, to signify completion of each of the four basic steps of MCCE: step1_out.pdb, step2_out.pdb, head3.lst, and pK.out.
pK.out signifies completion of step 4, so if a book.txt exists for a protein when mcce_stat is run, that protein will receive a "c" in book.txt to signify completion.
We recommend using mcce_stat with p_batch.
ms_hbond_percentages.py
Creates a table displaying all Hydrogen bond connections across microstate PDBs, and their percentages. Defaults to the local directory named pdb_output_mc_hbonds.
usage: ms_hbond_percentages.py [-h] [dir]
ms_top2pdbs
Stands for Tautomeric Charge MicroStates. Outputs: the top N tautomeric charge microstates, along with related properties energy (E), net charge (sum_crg), count, and occupancy (occ); a summary file identifying ionizble residues with non-canonical charge, and which residues that do not change charge over the topN set; and the top N files of each charge state in PDB and PQR format.
By default, charge microstates are retrieved at pH 7, and the number of most favorable charge microstates (N_TOP) returned is five.
usage: ms_top2pdbs inputpdb_filepath [-ph PH] [-n_top N_TOP]
p_batch (MCCE_bin)
Starts multiple protein runs at once, using the same set of instructions, and creates a book.txt file to manage their completion status. p_batch accepts a directory containing protein files, and (optionally) a shell script given custom instructions. If a shell script is not provided, a default one will be created, and may be edited to the user's preference. If a file named "run.prm.custom" is in the present working directory at runtime, the file will be read to override the default run.prm instructions.
p_batch creates a run directory for each protein file, and begins running MCCE for each one. Files will be created for their respective directories as each step is completed. Use mcce_stat to check how each run is progressing.
To stop a run in progress, delete the files or directory associated with the run.
p_info (MCCE_bin)
Gives a high-level summary of characteristics of a PDB file, including residue, chain, and ligand counts, as well as other aspects of a PDB changed during step 1 of MCCE, including how residues are named. If step 1 has not been run on the PDB file at runtime, p_info will automatically run step 1 before continuing as normal.
pdbs2pse (MCCE4-Tools)
usage: pdbs2pse file1.pdb file2.pdb ... [--pse_name <output_name>]
Converts one or more PDB files into a single PyMOL session file (.pse). The session file contains all the loaded PDB structures as separate objects. The user can specify an optional output name for the .pse file, or it will default to the name of the last input PDB file.
postrun (MCCE4-Tools)
usage (in a directory with sum_crg.out, pK.out files): postrun [-h] [-run_dir RUN_DIR] [--is_benchmark]
postrun provides basic diagnostics on sum_crg.out and pk.out files, after a run is completed. postrun looks for non-canonically charged residues, residues without curve fit or a chi-squared above 3, and residues that are out-of-bounds. The problem residues are outputted to the terminal and saved to a "postrun.bad" file. If there are no problem residues, a "postrun.ok" file is created instead.
postrun can be run on a directory of completed protein runs, with the flag "-run_dir".
txt_to_csv
A quick script that copies a given file into a .csv format. The source file does not need to be a .txt file. Recommended to use with spreadsheets.